2022年4月26日改訂
(1) 最新の構造決定の方法は。(分析化学)
(1a) 単離精製した後、概ね次の手順に従う。元素分析により組成式を知る。質量分析法で分子量や分子式を知る(IHDもわかる)。赤外分光法でどんな官能基があるかを知る。核磁気共鳴(NMR)法で炭素骨格の構造や官能基の位置に関する情報を得て、構造をほぼ確定する。よい単結晶が得られれば、単結晶X線回折法で構造を確定できる場合もある。構造が複雑な天然物の場合には、それでも論文で報告された構造が後になって誤りであったことが明らかになることがある。概要は「機器分析学」「環境計量学」などで教わる(2009年カリキュラム)。(1.1) 構造決定の歴史は?
(1.1a) 現在と同じような意味で有機化合物の構造が考えられるようになるには、まず 20世紀の前半には、新しい有機化合物の構造を決定するには化学的方法、すなわち確実な化学反応を用いて既知の化合物へと誘導し、得られた既知化合物の構造に基づいて、構造から反応前の構造を推定するしか方法がなかった。
このために、分子式が確定したら、
(1)官能基の確認
(2)炭素数の少ない(複数の)簡単な化合物へと誘導(減成反応という)
(3)生成物が構造既知の化合物かどうかの判定
(4)構造未知の化合物が見られなくなるまで(1)〜(3)の繰り返し
(5)同定された既知化合物の構造と(1)〜(3)の内容から、新化合物の構造を推定
という過程を経て、新化合物の構造が推定された。
並行して、19世紀後半〜20世紀前半には(1)、(2)の段階で用いられる化学反応が盛んに研究された。
(3)の段階では、得られた全ての化合物について分離精製と分子式の確定(元素分析および分子量の決定)が必要であり、手間と時間がかかった。さらに、(5)の段階では複数の可能な構造が考えられることも少なくなく、可能性の見落としもあれば、同じ化合物の構造をめぐる研究者間の論争もあった。
当時から、構造を最終的に確認するためには、
(6)既知化合物から出発して、確実な合成反応を用いて推定された構造をもつ化合物を合成し、問題の化合物と同じ物質であることを確かめる
必要があると考えられていたが、比較的簡単な天然物であっても、これが可能になったのは20世紀の中頃以降だった(代表的先駆者:R.
Robinson、R. B. Woodwardら)。(1)〜(5)の方法で推定されていた構造が、この段階で違っていることがわかった例も少なくない。
20世紀の後半になると、分子量の決定には質量分析法が有力な手段になった。また、紫外線吸収スペクトル、赤外線吸収スペクトル、そして1970年代には核磁気共鳴法などの分光学の活用法が確立され、分子構造との関係についての情報が蓄積されて、化学的方法による(1)〜(5)の段階に取って代わり、現在に至っている。元素分析法もガスクロマトグラフィーのような機器分析法を取り入れて進歩し、簡便になった。しかし、不安定な化合物の場合には、現在でも化学反応を用いて安定な化合物に変換してから構造決定に取り組む必要がある。
また、X線結晶解析法の進歩と共に、分子量のやや大きな化合物まで結晶構造が原子レベルの分解能で解析できるようになっている。それでも得られる構造が推定構造であり、時として論争や訂正が起こる場合がある点に変わりはない。
(2) 遷移元素は有機化学に含まれないのか。遷移元素の関係する有機化合物は一つもないのか。
(2a) 「有機金属化学」という1960年代に盛んになり現在も活発に研究が行われている分野では、遷移元素を含む有機化合物が重要な研究対象の一つである。(3) ATPは有機物か。
(3a) 有機物である。(4) 最近のシュガーレス飲料に含まれているマルチトースという糖の構造は?またなぜ体に吸収されにくいのか。
(4a) 「マンニトール」の間違いではないか確認して下さい。構造は図書室の「化学辞典」などに載っています。糖ではなく糖アルコールであることが吸収されにくい理由と関係があると思われます。甘いからといって必ずしも糖とは限りません。(5) どうして炭素が含まれるものが有機物質なのか?また、例外があるのはどうしてか?
(5a) これは定義ですので、「どうして」と聞かれても答えられません。ただし、定義されるにあたってはそれなりの歴史的背景があり、それを初回の講義で説明しました。生気論が信じられていた当時、有機物と考えられていた物質は例外なく炭素を含んでいました。また、当時から炭素の化合物で無機物とされていたもののほとんどは、現在でも「例外」として無機化合物として扱われています。(7) CO2のように炭素を含むのに有機化合物でない例外があるのはどうしてか?
(7a) CO2やCOの性質は、周期表で周囲にある元素の酸化物と比較して考えるほうが理解しやすいので、CO2と金属イオンからできる炭酸塩と共に、無機化合物として扱っています。HCNやシアン化物についても同様です。(9) 炭酸、塩酸、サリチル酸などはイオン性か分子性か?電離度との関係は?
(9a) 炭酸や亜硫酸は仮想の物質で実在しないので、この課題の対象外です。サリチル酸、酢酸、硫酸、塩化水素などは、純物質の状態ではほとんど解離していませんので、分子性物質です。塩酸は混合物(塩化水素の水溶液)であり、やはりこの課題の対象外です。また、電離度は水に溶けたときの性質ですので、この課題では基準になりません。
性質 |
分子性物質 |
高分子物質 |
分子式 |
明確 |
組成しかわからない |
分子量 |
小さく一定 |
大きく(概ね1万以上)不定 |
融点、沸点 |
低く一定 |
高いか不定、あるいは融解前に分解する |
水溶液(溶ける場合) |
溶液 |
コロイド溶液 |
(13) 人が初めて作った有機化合物が尿素なのはなぜか?尿素が一番作りやすかったのか?
(13a) 偶然です。生気論にとらわれていた当時の有機化学者は無機化合物から有機化合物を作ろうなどとは考えていませんでした。ウェーラーはもともと無機化学者であり、無機化学でも優れた業績を上げています。異性体の概念の確立も無機化学での彼の業績の一つでした。当時彼はたまたまシアン酸アンモニウムという尿素の異性体に相当する組成をもつ物質を合成しようとしていたのです。得られた変な物質を見て、異性体の尿素かも知れない(もしそうなら一大事だ)と考えたのもそんな彼だからこそかも知れません。そして、(生気論にとらわれて「そんなはずはない」と捨てないで)思いついたことを丹念に調べて尿素に違いないことを確かめた点で、ウェーラーは偉大な化学者だったのです。(14) 新しい物質ができたときに毒性があるとか、頭痛に効くとかどうしてわかるのか?
(14a) いずれもまず動物実験で調べます。頭痛に効く可能性が認められた場合は、動物実験で毒性や副作用がないことを確かめてから、試験的に人に投与して効力を確認します。(20) 有機化合物の毒性は実験してみないとわからないのか、それとも構造からわかるのか?
(20a) ある有機化合物の構造がわかっても、それだけでその化合物の毒性がどの程度かを知ることはできません。しかし、現在では多くの有機化合物の毒性データが知られており、それに基づいて構造と毒性との間にさまざまの経験則が見出されています。それらを利用すれば、新しい化合物の毒性がある程度予測できる場合も少なくありません。ただし、その予測がどの程度実態にあっているかは調べてみないと何とも言えません。この予測法につながる基礎的内容は、「分子設計」の中で学びます。(23)レシチン(リン脂質を含む脂質製品の総称)は分類できますか?
(23a)総称(集合名)の回答は,レポートの意図ではありませんが、しばしば見られます。また、総称であっても分類することはできます。レシチンは一般に「有機物ーイオン性物質」に分類されます。
[命名法]
(1a) 命名法は覚えるものではなく、規則を参照して構造に対応する名前を付けたり、名前に対応する構造を描いたりするものです。テキストやマクマリーの命名法の問題にあたっているうちに、身に付くはずです。(2) テスト等ではIUPAC名と慣用名のどちらを使ってもいいか。
(2a) IUPACが認めている慣用名(講義で括弧なしで使う)以外は使わないほうがいい。(6) IUPAC命名法は英語で書けないといけないのか?アルファベットで書く方がカタカナよりもいいのか?
(6a) 一つの名前の中で統一されていれば、日本語でも英語でも構いません。アルファベットで書くのは国際共通の方法です。しかし、IUPACは、各国語の文字や語法を用いて表記することを認めています。したがって、日本語の文章の中ではカタカナや漢字を用いるほうがいいのです。逆に、国内では、単に英語の綴りをカタカナにしただけの用語は誤りの場合もあります。例:○酢酸エチル、×エチルアセテート。○塩化アセチル、×アセチルクロリド。(7) ![]() の主鎖は、右上から枝分かれを下へ向かう部分(CH3CH2C=CHCH2OH)ではないのか(このほうが炭素数が多い)?
の主鎖は、右上から枝分かれを下へ向かう部分(CH3CH2C=CHCH2OH)ではないのか(このほうが炭素数が多い)?
(9) 物質名をつけるときに、「イソ-」を付けるときと付けないときがあるのは?
(9a) 「イソ-」などの接頭辞を付けるのは慣用的な命名法で、直鎖(ノルマル、n-)のものの異性体を指しています。炭化水素の名前では、IUPACでは他に置換基を持たない炭化水素に限って「イソブタン」「イソペンタン」「ネオペンタン」「イソヘキサン、(CH3)2CHCH2CH2CH3」の名前を使うことを認めています。また、基の名前では置換基がない場合に限って「イソプロピル」「イソブチル」「s-ブチル」「t-ブチル」「イソペンチル」「ネオペンチル」「t-ペンチル」「イソヘキシル」を使うことを認めています。(10) CH3CHClCHBrCH3のIUPAC名は2-ブロモ-3-クロロブタンと3-ブロモ-2-クロロブタンのどちらが正しい?
(10a) 2-ブロモ-3-クロロブタンが正しい。主鎖のどちらから位置番号を付けても数字が同じになるときは、より先に表される置換基の位置番号が小さくなるようにする(参考:平山ら「有機化学・生化学命名法 上」改訂第2版、南江堂、1988年、p. 13, A-2.4)。(12)
C4H6の異性体の一つ、![]() のIUPAC名を教えて下さい。
のIUPAC名を教えて下さい。
(13)「果糖」を「フルクトース」と呼ぶときがあるが、これはどうしてか。
(13a)「果糖」は漢語(日本語の一種)慣用名で、「フルクトース」は欧米語(慣用名)の綴りや発音から作られた外来語(カタカナ語、これも日本語の一種)です。糖類などの生物体に由来する物質は慣用名を用います。慣用名ですので、どちらが正式ということはありません。どちらも広く使われています。漢語の慣用名は糖では「果糖」の他に単糖類では「ブドウ(葡萄)糖(グルコース)」、二糖類では「ショ(蔗)糖(スクロース)」「麦芽糖(マルトース)」「乳糖(ラクトース)」、多糖類の「澱粉(でんぷん)」がよく使われます。(14)有機物の名前からおおよその機能を推測することはできるか。
(14a)名前から官能基を読み取ることができれば、その官能基に特有の性質をもつことが予測されます。IUPAC(推奨)名は構造式と対応していますので、名前から構造式を描くことができれば、さらに多くの性質を推測することができます。[結合と構造]
(1a) 弱い結合がある場合と、構造にひずみがある場合とがある。(7) 分子式からIHDがわかったあと、構造式を書くにはどうすればいいか。特にIHDが小数の場合。
(7a) 方法は多数あります。IHDを何に充てるか、どのような炭素骨格を持つか、どのような官能基をもつかを考えるのが一般的な方法です。IHDが小数の場合は、たとえば、水素を一個追加して「中性分子」を作り、できたものから水素を一個取り除くというのも一つの方法です。(10) C−O−O−H(C)という結合は不安定だとすると、どうなるのか。
(10a) 少し加熱したり、紫外線を当てると容易にC−O・と・O−H(C)に分解します。しかし、周囲にこれと反応しやすい分子がなければ、再び結合することもあります(可逆反応)。(14) 異性体を書くのに決まりごと(あってはならない構造)はないのか?
(14a) 有機化学では、共有結合の原子価が不完全なものは考えません(無機化学では、CO、NOなどのように、これに該当する分子が登場します)。5価の炭素など、価電子殻では収容できない数の価電子をもつ構造は、当然、誤りになります。(15) IHDに対応する不飽和結合はCとOの間(Cと他の原子の間)でも考えられるのか?
(15a) いい質問です。考えられます。Cに限らず、ヘテロ原子間(NとN、NとOなど)でも考えられます。(16) 構造異性体を書くとき、配座異性体も考えられれば書くのか?
(16a) 配座異性体は構造異性体ではないので、特に指示がない限り書く必要はない。立体(配置)異性体も同じ。(18) 炭素は4価なのに(丸善HGS)分子模型の炭素の「たま」には穴が5つあいているものがあるのはなぜか?
(18a) その「たま」はsp2混成やsp混成の炭素原子を表すために用います。互いに120°になっている3つの穴を用いると、sp2混成の炭素原子を表すことができます。またこれらと直交している2つの穴は互いに180°になっており、sp混成の炭素原子を表すことができます。(19) メチル基が左側にあるときに、CH3-と描く場合とH3C-と描く場合があるが、どちらが望ましいか。
(19a) どちらでも構いません。それぞれに根拠があります。前者は「分子の骨格をなす原子に結合している原子(団)は、骨格原子の右に書く」という(示性式の)原則に従っています。一方、後者は「結合を表す−の隣りには、結合している原子を描くという(完全な構造式の)原則に従っています。 CH3-を用いる表記は両者の中間にあるので、両方の表記が可能です。また、このことから、右側にあるときに -H3Cはどうして不都合かがわかると思います。(22) Na、K、CaなどとCとの化学結合が不安定なのはどうしてか?
(22a) Na、K、Caなど電気陰性度の小さい原子(陽性の原子)とCとは、電気陰性度の差が大きいので、イオン結合を作ろうとする。しかし、一般に有機化合物中の炭化水素鎖を構成するC原子は安定な陰イオンにならないので、これらの原子とは安定な(イオン)結合を作らない。(23) 「元素組成から分子式を区別する」とはどういうことか。
(23a) 分子式があれば元素の質量組成を計算することができます(計算値)。一方、分子式が未知の物質を構成する元素の質量組成は実験で求めることができますが(実験値)、これには必ず誤差が伴います(設問では誤差範囲をプラスマイナス0.5%とした)。誤差範囲を考えに入れたときに、得られた実験値に基づいて推定できる分子式が一つになるとは限りません。二つ以上の分子式から与えられる元素組成の計算値が、得られた実験値と誤差範囲で一致するとき、これらの分子式は互いに区別することができないことになります。(24) 配位結合は英語でcoordinate bondか、dative bondか?(重要)
(24a) 結論からいうと「どちらも正しい」です。ただし、両者は内容が異なりますので、伝えたい内容に応じて一方を選ぶ必要があります。重要なことですので、その根拠を少し詳しく説明します。 まず、あまりなじみのない dative bond の意味を、IUPAC(国際純正および応用化学連合)のホームページにある用語解説のページ(通称:Gold book)から転載します。[電子状態]
(1) σ結合の分子オービタルの図(エネルギー図)は何を表しているのか。
(1a) 原子オービタルと分子オービタルのエネルギー準位の相対的な上下関係を表しています。(4) 炭素原子の混成状態について、二重結合があるからsp2というわけではないとしたら、sp2かspかはどうやって見極めるのか?
(4a) 炭素原子が直接結合している原子の数(単結合か多重結合かは無関係)で決まります。4個ならsp3混成、3個ならsp2混成、2個ならsp混成です。
[炭化水素]
(1) シクロアルカンが平面・ほぼ平面・非平面のいずれかを判断する時、考慮するのは炭素原子のみでよいのか?
(1a) 環を構成している炭素原子に注目して判断する。一般に環状化合物の構造を考えるときには、環を構成する原子にだけ注目して判断すればいい。(2) 二置換シクロアルカンの場合にcisーtrans異性体があるのなら、三置換体以上の場合はどうなるのか?
(2a) どんな置換基がどこについているかによってさまざまの構造異性体が存在する。さらに、各構造異性体について2つ以上の立体異性体が存在する。立体異性体は多種多様なので、二置換のときのような慣用名は用いない。(3) なぜシクロアルカンでは位置異性体をオルト、パラ、メタで呼び分けないのか?
(3a) 二置換ベンゼンの位置異性体は三種類だけであり、それぞれ慣用名のオルト、パラ、メタで呼び分けます。しかし、二置換シクロヘキサンの位置異性体はずっとたくさんあります(分子模型を組んでみればわかる)。位置異性体の他に、たとえば、1,2- 二置換の異性体(ベンゼンではオルト異性体にあたる)には少なくとも2通りあります。3つの慣用名だけではどの異性体をさすのかを明確に示すことができないので、かえって混乱します。おそらくこれが用いられない理由でしょう。(4) シクロヘキサンにはイス型と舟型があり、一般にはイス型が安定であるが、両者は異性体ではないのか?
(4a) 両者は同じの分子のもつ2つの「形」であって、互いに入れ替われるので、異性体ではありません。「立体化学」で詳しく取り扱います。[共役系と電子の非局在化]
(1) 電荷の非局在化というのはどういうことが起こっているのか。また、どのような場合に起こっているのか。
(1a) 電荷が特定の原子の上だけに孤立して存在するのではなく、複数の原子上に分散している状態を「電荷の非局在化」といいます。(2) π電子の非局在化は二重結合が二つあるときしか起こらないのか?
(2a) 二重結合または三重結合、そしてこれと陽イオン(空の2p軌道)、陰イオン(電子対の入った2p軌道)とが隣接していても(共役系)起こります。(5) 共役π電子系で「二つのπ結合の間」(非局在化しなければ単結合であるところ)のπ電子は左右どっちの電子か見分けるのは本当にできないのか?
(5a) できません。一個の電子の位置を正確に定めることはできません(不確定性原理)。また、炭化水素系では、左右のπ電子が「二つのπ結合の間」に非局在化する確率は等しいですので、どちらの電子のほうが「二つのπ結合の間」にある可能性が大きいともいえません。(6) 電子が非局在化して安定化するとき、その前の形から変化するのに余分のエネルギー(活性化エネルギーのことか?)は必要ないのか?
(6a) 非局在化しているとは、「局在化した状態から非局在化した状態へ変化する」ということではなく、「古典的な結合理論に基づく構造式では局在化しているように思われる電子が、実際には構造式から予想されるのとは違って、特定の原子間を超えて拡がっている」状態を指します。「変化する」と考えるのは前提が間違っています。(8) 限界構造式を描くときに、どの部分にまず注目すればいいか?
(8a) 電荷をもつ原子、非共有電子対や(あまりないが)空のp軌道をもつ原子、極性のある二重結合、極性のない(C=C)二重結合の順に注目します。(9) 限界構造式で考えるときに、上下や左右をひっくり返せば全く同じ構造を2つ描くのはなぜか?
(9a) ひっくり返せば全く同じ構造になるのは、分子の対称性がいいからです。しかし、同じ原子に注目すると電子の局在化の様子が違いますので、限界構造式としては同一ではありません。そのため、片方を省略すると、電子の非局在化の様子を正しく表現することはできなくなります。分子の対称性がいい場合には、このことを逆に利用して、限界構造式を抜かりなく探すことができます。(10) 結合の長さの表で、sp2混成のC−C(単)結合の長さとC=C(二重)結合の長さの違いは何によるのか?
(10a) π電子の非局在化の程度の違いによるものです。ベンゼンのような場合を除いて、π電子は共役系全体にまったく均等に非局在化しているわけではなく、π電子が少し少なくなった二重結合と、少しπ電子が入ってきた単結合が交互に並んでいるのです。(11) ブタジエンやベンゼンなど炭化水素の非局在化を考えるとき、対イオン(「プラスとマイナスに分かれている」もの)ではなく、不対電子が2つあるような限界構造式は考えないのですか。
(11a) 不対電子が2つあるもので考えても構いません。しかし、このような問題で電子が非局在化する範囲を考える時には、対イオン(「プラスとマイナスに分かれている」もの)で考えても同じ結論が導き出せます。そこで、普通は対イオンで考えます(やや高度な内容なのでこの授業では扱いませんが、重要な概念である「芳香族性」を理解するには対イオンのほうが適しています)。(12)上の質問で、対イオンでは分子内にプラスとマイナスの電荷が分かれて存在していて、不安定ではないのですか。
(12a) 対イオンは、電子対が局在化している分だけπ電子(非局在化している)の数が少なくなりますので、もとの構造に比べると不安定と考えられます(どちらも現実には存在していない構造なので実際のところはわからない)が、正負の電荷が隣接していない構造は、限界構造式を用いて非局在化を考えるときには考慮に入れます。ブタジエンと同様に、そうしてはじめてこの化合物でのπ電子の非局在化が理解できます。(13) ベンゼン環を描くときに、六角形の中に○を描く方法でもいいのか。
(13a) それで構いません。しかし、炭素の原子価を把握したり、「有機化学(2)」で学ぶ「電荷の非局在化」を限界構造式を用いて考えるには、ケクレが提案した単結合と二重結合を交互に描く構造式のほうがわかりやすいので、ほとんどの化学者はケクレ構造で描き表します。しかし、それによって「6つのC−C結合はまったく同じ長さであり、区別できない」というベンゼン環の実態が変わるわけではありません。[構造と性質の関係、水素結合]
(1a) 分子間力が大きくなるので、大きなエネルギーを与えないと液体の状態にある分子同士を互いに引き離すことができないからです。(2) 沸点の高低を考えるときに極性と水素結合はどちらを優先して考えればいいか?
(2a) 一般的には水素結合のほうが分子間力としては強いですが、極性の大きな分子では、弱い水素結合に匹敵するほどの分子間力を示す場合があります。(3)(たとえばカルボキシル基の)一個の酸素原子が二個のドナー水素と水素結合することは絶対にあり得ないのか?
(3a) あり得ます。分子は多数あるので、中には二個の水素原子と水素結合しているものもあれば、まったく水素結合していないものもあります。また、液体では常に動き回っていますので、時間と共に水素結合の様子も変化しています。図示しているのは平均的な様子です。(4) 極性の大きい分子、水素結合している分子が水に溶けやすいのはなぜか?
(4a) 極性の大きい分子は、水分子に取り囲まれると極性分子間の相互作用を起こすからです。これを一般に「溶媒和」特に水の場合「水和」といいます。水分子は特に水素結合しやすいので、水素結合する分子は水中では周りを取り囲んだ水分子と水素結合して安定化します。このため水に溶けやすいのです。(5) 構造異性体を式で書くときに、考えられても書いてはいけない構造はあるか?
(5a) 同一の構造に対応するものを2つ以上書くこと(試験では、同一性を見分ける問題でなければ減点対象にはならないが、同じものを2回数えた結果として、書くべき構造を見落としたら減点される)。原子価が間違っているもの。(6) 有機化合物の炭素原子に結合した水素原子を他の原子に変えたら、形や結合角は変わるのか?
(6a) 単結合は速やかに回転できますが、第6章で学ぶ安定な配座が変わるために、形が変わる場合があります。また、結合の長さや結合角は、最適の長さや角度を基準として、常に振動していますので、分子内の原子の相対的な位置はいつも一定なわけではありません。いいかえれば、(単原子分子以外の)分子は結合の振動や回転のために、常に形を変えているのです。炭素原子に結合している原子が変わると、結合の長さが変わるために、分子内の他の原子とのぶつかり合いが生じ、そのために最適な長さや角度が変わることはよく起こります。(1) 立体化学がよくわからないので、安い参考書を教えて下さい。
(1a) 「シラバス」とホームページに例を挙げています。題名に「立体化学」と書かれている本ならたいてい、一回の飲み代か七日分のタバコ代でおつりが来ます。「日本理学書総目録」(創学サービスの注文カウンターなどにあるはず)を参照して下さい。(3) どのようなときにE、Zを使い、どのようなときにcis、trans を使うのか。
(3a) E、Zはどのような場合にも使えます。IUPACの勧告にも沿っていますので、こちらを使うのがいいと思います。cis、trans を使うときは、注目する置換基(2つ)を指定するのが原則です。ただし、アルケンの各炭素の水素原子を一つずつ他の置換基で置換した場合(1,2-二置換アルケン)には、注目する置換基は自明ですので、指定しないで使われます。(4) 「幾何異性体」は「シス・トランス異性体」が正しいのか?
(4a) どちらも誤りではありません。IUPACの立体化学用語法(Basic Terminology of Stereochemistry)では、一時、「幾何(geometrical)異性(体)」という用語を使わないで「シス・トランス(cis-trans)異性(体)」を使う方向の勧告が出たことがあります(1996年の勧告)。しかし、その後、錯体化学で取り扱う幾何異性の中に「シス・トランス異性」の定義では分類できない(他の「立体異性」にも分類できない)ものがあり、「幾何異性(体)」の名称を残さないと不都合のあることが指摘されています。また、cis-, trans- の表記を使わないで E, Z を用いることを推奨しながら、異性体の分類では用いるという矛盾に対する指摘もあります。このため、最近では「幾何異性(体)」の名称を残す方向で検討がなされています。(6) 配座異性体のエネルギー差はどのようにして求めればいいのか。
(5a) 各異性体の存在比が実験的にわかれば、それから求めることができます。実験的に観測が難しい場合には、理論化学的な計算によって推定します。(8) 置換シクロヘキサンの立体構造で、cisとtransはどうやって区別すればいいか。
(8a) 炭素骨格が平面六角形だと仮定すると、各炭素原子の置換基は平面の上と下に一つずつあるはず。このとき、上(下)にある置換基同士の関係がcis、上にある置換基と下にある置換基の関係がtransになる(環反転が起こってもこの関係は変わらない)。平面に近づくように、分子模型を少しひねってみればわかる(ひねりすぎると結合が折れるので注意すること)。(9) ひずみエネルギーとは?どのようにして生じるのか?(化学結合論)
(9a) 共有結合には、それぞれ最適の長さ(結合長)や結合間の角度(結合角)があります。実際の分子の結合長や結合角が、最適の大きさからずれる(これを「ひずみが生じる」または「ひずむ」という)には、それだけのエネルギーを与える必要があります。ひずみが生じるために必要なエネルギーをひずみエネルギーといいます。(11) ねじれ形と重なり形では、なぜねじれ形のほうが安定なのか?
(11a) ブタンの(C2-C3間の)ねじれ角 0° の重なり形のように、両端のメチル基が近づきすぎるような場合には、重なり形が明らかに立体的に不利であることは理解できると思います。しかし、エタンではどうでしょうか?重なり形のときに水素原子どうしが立体的に近づきすぎているとは考えられません。別の理由があるはずです。この場合に重なり形が不安定なのは、隣接する炭素原子でC-H結合を作っている共有電子対どうしが、重なり形のほうが近接しているため、クーロン反発がねじれ形よりも大きいことによると考えられています。(3) Fischer投影式から立体構造を想像できるようになるにはどうすればいいですか。
(3a) 分子模型を使って慣れることがもっとも効果的です。(6) 絶対配置のR(時計回り)・S(反時計回り)は、旋光性の+(時計回り)−(反時計回り)と対応しているのか?
(6a) 対応していません。両者の間には論理的・経験的な関係はありません。(7) 旋光性の回転方向はどのように判断(予測)すればいいのか?
(7a) 構造式や立体構造から旋光性を説明・予測できる簡単な理論や経験則は知られていません。(10) 結合がいくつもあるとき、どこを視点として優先順位を考えればいいのか?
(10a) 不斉炭素原子から出ている4つの結合に注目します。(11) 環を構成する異なる(炭素)原子にそれぞれ1つずつ置換基が結合したときにできる立体異性体は幾何異性体か、それとも光学異性体か?
(11a) どちらでも正しいです。異性体を個々に区別するときには、一般により細かい区別のできる立体異性体の命名法を主に用います。(12) Fischer投影式で書く場合とそうでない時とではどういう違いがあるのか。また、それは何のためか?
(12a) Fischer投影式は、立体配置(Configuration)を化学式で表現するためのもっとも便利な方法です。しかし、反応を含めて、立体化学を理解するためには、必ずしも便利とは言えません。一つの理由は、立体配座(Conformation)の情報を持っていないからです。特に反応の立体化学を考える際には、立体配座を明記しないと理解できません。また、環状化合物の環にある不斉炭素の立体配置を示すにも不便な場合があります。(13) Fischer投影式の上下の置き方はどんな場合でも決まっているのか?
(13a) 糖やほとんどのアミノ酸では決まっていますが、あらゆる光学活性物質について上下が規定されているわけではありません。また、あくまでガイドラインであり、従わないと誤りというわけではありません。もちろん、糖関連物質などでは、従ったほうがコミュニケーションが行いやすくなります。[天然物有機化学]
(1) 化学辞典や国語辞典「精油」の項に載っていますので見ておいて下さい。
植物から取れる揮発性、特有の香りをもつ油状の液体。多くは水に溶けない。香水その他の香り成分として化粧品、せっけん、食料品などに用いる。化学的にはテルペン類と呼ばれるC10またはC15の各種化合物またはフェノール類が多い。
(2) 第一に、核磁気共鳴法(NMR)のような構造決定のための優れた手段が普及し、全合成を行うまでもなくたいていの構造を決めることが可能になったこと。第二に、よい単結晶が手に入り、X線結晶解析を行うことができれば、たいていの天然物の構造は間違いなく決定することができるようになったこと。この2つが大きな理由です。また、有機合成の技術が進歩し、複雑な化合物でも全合成できることがわかってきたために、(皮肉な話ですが)合成化学者にとって全合成が昔ほど魅力的な研究テーマではなくなったこともあります。それでも、NMRで決定的な結論が得られず、よい結晶が得られないときは、全合成で結論を出すことが今でもあります(時には提案されていた構造が全合成で覆ることもあります)。
(3) 天然物のような複雑な有機化合物を、構成する元素の単体から出発して、化学反応を組み合わせて合成すること。実際には多数の有機化合物が単体から合成できることがわかっているので、そのような有機化合物を出発物質として合成する。
天然物の分子構造を確認する手段であると同時に、有機合成化学の限界に挑戦してその可能性の幅を広げる研究でもある。
(4) あります。酢酸カーミンは、カーミン(carmine)という色素の酢酸水溶液です。この色素の正体はcarminic acidつまりカルミン酸です。
(5) 私たちの体そのものが有機物であり、成長し、また体を維持するためには有機物が必要です。また呼吸により有機物を酸化して、生きていくためのエネルギーを手に入れています。このために多種多様な有機物を体内に取り込んでいます。その一方で、酸素、水、食塩やその他のミネラルなどの無機物も、実際にはたくさん取り込んでいることも見逃せません。
(6) 藍の成分である藍色の染料はインジゴと呼ばれ、下記のような構造をしています。
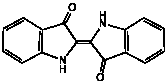
貝紫の成分である6,6'-ジブロモインジゴと同じ炭素骨格をもっています。
[電子効果]
A1. いい質問です。アルキル基が電子供与基に分類されるのは、(1)陽イオンを安定化させる、(2)陰イオンを不安定化させる(授業の範囲では出てこない)、(3)アルキルベンゼンの双極子モーメント、(4)アルキルベンゼンの求電子置換反応での反応性などの振る舞いが、電子供与基のもつ性質と共通しているからです。
その理由はC-H結合の極性のためにCの電子密度が大きくなり、やや負に帯電しているためだとするのが一般的ですが、他にもいくつかの説明があります。簡単なようでいてもっとも難しいのが炭化水素基の振る舞いです。
生体内でも、核酸塩基にメチル基がつくと、その挙動がまったく変わることが知られています。
A2. します。酸素原子のもつ非共有電子対は、隣接するπ電子系と同一平面になることができるので、共役系を構成し、π電子系の方へと非局在化することができます(電子供与性の非局在化効果)。この様子を表すために、酸素の非共有電子対をπ電子系のほうに移動させたような限界構造式を描きます。
一方、カルボニル基の酸素の非共有電子対は、π電子系と同一平面にはありませんので、π電子系のに非局在化には関与しません。
こういうことも、分子模型で考えるとよくわかります。
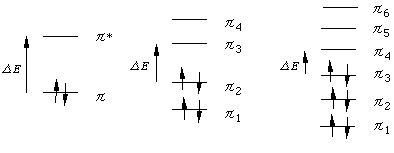
A3. π電子の非局在化が大きいと、分子の分子オービタルのエネルギー純位は下図のようになり、電子の入っている一番上の結合性オービタル(HOMO)と、電子の入っていない一番下の反結合性オービタル(LUMO)とのエネルギーの差ΔEが小さくなります。Planckの式
ΔE=hν=hc/λ
より、ΔEが小さくなると、分子により吸収される電磁波の波長λは大きくなります。
A4. 紫外線・可視光の吸収スペクトルは共役π電子系の電子状態の変化によって変わります。メチルオレンジはpHが小さくなると窒素原子の非共有電子対にプロトンが結合するため、この非共有電子対が共役系から外れます。この電子状態の変化によって、上の(3)に記したように吸収スペクトルが変化し、目に見える色が変わります。他の指示薬の変色も同じような原理によります。メチルレッドの構造は「化学辞典」などを見て下さい。
A5. 炭素陽イオン(carbocation)の中心炭素はsp2混成であり、2p軌道には電子が入っていません(下図(a))。炭素陰イオン(carbanion)の中心炭素は、共役系に含まれていない場合はsp3混成で、他の原子と結合していないsp3軌道に電子が2個入って非共有電子対となっています(下図(b))が、共役系に含まれている場合にはsp2混成となり、2p軌道に電子が2個入って非共有電子対となっています(下図(c))。この非共有電子対が共役系へと非局在化すると説明されています。

A6. あります。正電荷が共役系に非局在化すると、共役系は電子不足になり、求核種の攻撃を受けやすくなります。逆に負電荷が共役系に非局在化すると、共役系は電子過剰になり、求電子種の攻撃を受けやすくなります。
A7. それぞれの効果の特徴については第1回で説明しました。両者が同時に働くのは、根元にN原子やO原子をもついくつかの官能基の場合です。これらの原子の電気陰性度が炭素より大きいことからもわかるように、誘起効果では炭化水素骨格から電子を引っ張ります(陰性基)。一方、これらの原子が非共有電子対をもっていて、それが共役π電子系に接している場合には、A2に書いたようにこの電子を共役系に与えるように非局在化します(電子供与性の非局在化効果)。共役系では電子供与性の非局在化効果が大きな影響を及ぼすので、その場合、誘起効果を考慮する必要はありません。

A8. 電子の分布を考えるにはどちらか一方から考えれば十分ですので、理解に時間のかかる電子供与基のほうから説明しました。実際には両方の効果がベンゼン環に働いています。限界構造式をもれなく描くには、それぞれを考えた限界構造式をすべて描かなくてはなりませんが、これは面倒な作業ですので、大学院の試験でも求められることはありません。練習のつもりで、NO2基から考えて、残りの限界構造式を描いてみて下さい。電子の分布について同じ結論が得られるはずです。
A9. ほとんどすべての「有機化学」の教科書に必要最小限のことは書いてあり、その応用例は「酸と塩基」「芳香族求電子置換反応」「カルボニル化合物の反応」をはじめとして、あらゆる教科書全体にたくさんちりばめられています(「クラム有機化学」「パイン有機化学」「ソロモン有機化学」「マクマリー有機化学」など)。それらの説明ではよく理解できない場合には、クライン(竹内、山口訳)「困ったときの有機化学」(化学同人)(第2版では上巻)の「2章 共鳴」がいいでしょう。
(限界)構造式も非局在化の式も、有機化合物の実態(たとえば「電子の非局在化」)を理解するためのモデルです。優れたモデルほど実態をよりよく反映していますが、モデルは「真の実態」そのものではありません。(1)
ルールに沿って正しいモデルを作ると共に、(2) モデルと実態との関係を理解することが、モデルを通じて実態を理解する上で重要なことです。
A10. 「真の構造により近い」ということです。描かれたいくつかの限界構造式から真の構造(電子の非局在化している様子)を理解しようとするときには、「寄与の大きい」限界構造式に、より多くの重みをつけて考慮すればいいということです。
[酸と塩基]
A1. 水素結合は空間経由の相互作用なので、ドナーとアクセプターの位置関係によってその影響は異なってきます。水素結合が著しい影響を与える一例をテキストに紹介しました。
A2. 等しいように見えるのは偶然です。pKaの値についてはもう一桁詳しい報告もありますが、数値そのものや大小関係はそれほど重要ではないので、講義資料では丸めて表しています。
CH3置換体の挙動がp-NH2と同様である点については、FAQのページの「有機化学II関係」[電子効果]Q1.を参照してください。
A3. 過酢酸(CH3COOOH)のpKaは約8.2で、酢酸(pKa約4.7)よりも弱い酸であることがわかります。共役塩基の安定性を比べてみてください。
A4. 混成軌道の種類による形と広がりの違いが関係しています。同じ原子の作る混成軌道では、sp3混成軌道のときに、原子核からもっとも離れたところまで広がっているため、H+が配位結合しやすいからです。混成軌道は、sおよびpの原子軌道をもとにして作られているので、sp3混成軌道ではp軌道の性質が相対的に顕著に現れ、sp混成軌道では逆にs軌道の性質が顕著に現れます。s軌道の電子雲は原子核から比較的近い位置に分布し、p軌道の電子雲のほうが原子核から離れた位置に分布します。したがって、p軌道の性質が相対的に顕著に現れるsp3混成軌道の電子雲のほうが、原子核から離れた位置に分布します。混成軌道の「拡がり」については、このページを見て想像して下さい。
[有機化学反応概論]
A1. 詳しい機構は15章で扱います。アルコールのOH基と結合した炭素はsp3混成であり、カルボキシル基のOH基と結合した炭素はsp2混成です。一般にカルボニル炭素のほうがアルコールの付け根の炭素よりも求核種の攻撃を受けやすいのです。その理由は構造式と分子模型をよく検討すれば大体見当がつきます。
A2. 一般的には決まっていません。有機化学では、「有機化合物」に「試薬」が接近するという表現が一般的ですが、規則ではありません。ずっと重要なのは、「電子がどこから出てどこに行くか」です。
A3. H+は求電子種(E+)の一種です。一方、すべての求核種(Nu-)は、強さの差はあっても、同時に塩基(B-)でもあります。酸(H+)に向かって攻撃する場合に限って塩基(B-)と呼びます。
このような不整合性は、H+を相手に与えるものを酸と見なす Brφnsted の酸・塩基の定義に起因するものです。これに対して
G. N. Lewis は、電子対供与体を塩基、電子対受容体を酸と定義することにより、酸・塩基の概念を拡張しました。この定義によると、すべての求核種が同時に塩基であるのと同様に、すべての求電子種は酸であることになり、上のような区別は意味をなさなくなります。
A4. 次の手順で考えるとわかりやすいと思います。
まず、反応系に酸または塩基があるかどうかを判断する。次に、酸化剤・還元剤、そして求核種・求電子種が存在するかどうかを判断する。
次いで、これらがどこに作用するかを判断する。反応の起こりやすい位置(電荷、極性結合、非共有電子対、π電子)を判定する。また、適当な脱離基があるかどうかを判断する。
以上を基にして、何が起こるかを検討する。
詳細は2つ下のA6を参照して下さい。
A5. 多くの反応系では、反応の進行とは無関係のいくつもの化学平衡が生じています。反応経路は決して一本道ではありません。自分で反応を考えるときにうまくいかないのは、(a)反応の進行に必要な化学平衡に気がつかない場合(対策:見落としている経路がないか探す)と、(b)反応の進行に無関係な化学平衡にとらわれている場合(対策:袋小路に陥ったら戻って別の経路を進む)とがあります。
迷路を解くのに似ていますが、袋小路にはちゃんと目印があります。それが見分けられるようになれば、反応生成物に到達するのが容易になるでしょう。
A6. 概要は2つ上のA4と同じです。有機化学反応を一通り学んだ人のための詳しい説明を「まとめ 有機化学反応の考え方」(PDFファイル)にまとめました。
A7. 2つの異なる有機化合物R1-XとR2-Yから、より大きな(炭素数の多いI有機化合物R1-R2が生じる反応。
R1-X + R2-Y → R1-R2 + X-Y
置換基X、Yおよび用いる触媒を適切に組み合わせることによってうまくいくことがある。この組み合わせの発見が2010年のノーベル化学賞の対象となった業績。詳細はWikipedia等で調べて下さい。
A9. 「燃えにくい」という言葉はいろいろな視点から考える必要があります。以下ではハロゲンとして塩素を例として考えます。
(1)まず、酸化されにくい(酸素との反応が遅い)という視点から考えてみます。
炭化水素の酸化反応は、酸素原子やその他のフリーラジカル(遊離基)中間体によるC-H結合の水素引き抜き反応により進行する反応です。一般に水素原子はフリーラジカルによる引き抜き反応を受けやすいので、水素原子がすっかり酸素原子に置き換わって、二酸化炭素と水になるまで進みます。一方、C-Cl結合では、これと対応する塩素原子の引き抜き反応は非常に起こりにくいのです。・OHラジカルによる水素引き抜き反応の活性化エネルギーの例を下の表に示します。四塩化炭素からの塩素原子の引き抜き反応の活性化エネルギーが大きいことがわかります。
|
|
/kJ mol-1* |
| ・OH + CH4 → H2O + ・CH3 |
|
| ・OH + C2H6 → H2O + ・CH3 |
|
| ・OH + CCl4 → HOCl + ・CCl3 |
|
(2)燃え続けるためには、反応によって熱が供給される必要があります。
ここでは、切断されるC-H結合とC-Cl結合および生成するO-H結合とO-Cl結合に着目して、結合解離エネルギーを用いて反応熱を比較してみます。下の表は、関係する各結合の結合解離エネルギーをまとめたものです。H-C結合がH-O結合に組み変わる反応は多少なりとも発熱反応であるのに対して、Cl-C結合がCl-O結合に組み変わる反応は明らかに吸熱反応であることがわかります。このことからも、ハロゲン化炭化水素のほうが燃焼には不利であることがわかります。
|
|
/kJ mol-1* |
|
|
|
|
|
|
|
|
|
|
|
|
以上の結果は、ハロゲン化炭化水素自身が完全燃焼しにくいことを示しています。
ハロゲン化炭化水素自身が燃えにくいことはわかったと思いますが、これらはC-H結合をもっていて酸素との反応は起こり、さまざまのハロゲン化合物が生じますので、消火剤に用いるには向いていません。そこで、消火剤にはC-H結合をもたないハロゲン化炭素(四塩化炭素、ハロンなど)が使われます。これらの化合物は、上記と同様の理由からきわめて難燃性であることに加えて、次のような性質をもっています。
この結果、多くの初期消火の現場で
という燃焼に必要な三要素を分断することができますので、消火剤として好適なのです。
[求核置換反応]
A1. これらは反応をその機構で分類するための記号です。SはSubstitution(置換反応)、NはNucleophilic(求核的な)のそれぞれ頭文字から取ったものです。1や2の数字は、律速段階に関与する分子数を示します。したがって、SN1は一分子求核置換反応、SN2は二分子求核置換反応を表します。さまざまなタイプの反応に対応する記号が用意されていますが、専門家以外でも知っている必要があるのは、SNの他にはE(Elimination、脱離)とE(Electrophilic、求電子的な)くらいです。
A2. 反応機構が違います。
SN1では、反応物からまず脱離基が取れて炭素陽イオンが生成します。この段階が律速段階です。生じた炭素陽イオンに、求核種は速やかに反応するので、反応速度は求核種の種類や濃度が変わっても変化しません。第三級の炭素に脱離基が結合している場合など、安定な炭素陽イオンが生じる場合にはこの機構で求核置換反応が起こります。
SN2は、安定な陽イオンが生じない場合に起こります。反応物に求核種が攻撃すると同時に、脱離基が取れて生成物が生じます。したがって、律速段階には反応物と求核種の両方が関与します(二分子反応)。したがって、反応速度は、反応物の濃度だけでなく、求核種の種類や濃度によって変化します。第一級の炭素に脱離基が結合している場合などは、通常はこの機構で求核置換反応が起こります。
第二級の炭素に脱離基がついている場合には、求核種や反応条件によってどちらか一方だけが進行したり、両方が同時に進行したりします。一般に、強い求核種の場合ほどSN2が起こりやすく、弱い求核種の場合には(SN2が起こり難いため)相対的にSN1が起こりやすくなる傾向があります。
A3. 暗記するのは別に構いません。しかし、大切なのは各反応機構の本質(どのようにして起こるか、どのような場合に有利か)を理解することです。表はそれを簡潔に整理したものでしかありません。理解なしにただ暗記しても、判断するのには役に立たないでしょう。
理解の助けとなるQ&Aが上下にあります。のぞいてみて下さい。
A4. CH3CH2OHのC-O結合は、NaOHの場合と異なり、共有結合です(しかもC-Cより強い)。OH基は弱い脱離基なので、CH3CH2OHなどからひとりでにOH-が脱離することはありません(だからエタノール水溶液は中性ですし、有害なOH-の生成を心配することなく、成人はこれを飲んでその生理作用を楽しむことができるのです)。C-O結合が切れるのは、(2)のように、酸性条件下でプロトン化して−OH2+(=よい脱離基)となり、OH2(水)として脱離する場合がほとんどです。OR基も同じです。
[脱離反応]
A1. β位の水素がとれやすいのは、テキストに示したような自然な電子の流れが起こり、二重結合ができるからです。この講義では扱いませんが、α脱離もあります(下図)。α脱離は、β脱離が不可能な場合に、より強塩基性の条件下で起こります。弱い酸性を示すCHがH+として塩基により奪われて反応が開始する多段階反応であり、β脱離とは反応機構が異なります。
脱離基に対して(β位でなく)γ位の水素が酸性の場合に塩基性条件下で起こり、三員環を生じる環化反応をγ脱離と呼ぶことがあります。こちらも、弱い酸性を示CHがH+として塩基により奪われて反応が開始する多段階反応です。脱離反応ではなく分子内の求核置換反応(SN2反応)と考えるのが普通です。
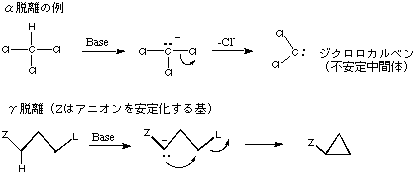
A2. もしこの結合が切れるとすると、不安定なCH3+イオンが生成することになります。また、生成したCH3+イオンからは、アルケンのような安定な生成物はできようがありません。
[付加反応]
A1. RO・は、HBrの水素原子と反応して(「水素引き抜き」という)ROHとなり、Br・が生じます。Brと反応しないのは、O-H結合のほうがO-Br結合より安定なためだと説明されています。できたBr・がアルケンのπ電子を攻撃してBrCH2-CH2・ができ、今度はこれがHBrの水素原子と反応します(ラジカル連鎖反応)。したがって、このような機構で反応が進む限り、H・は生成しません。
A2. まだよくわかっていません。プリントに書いてある酸素が3つならんだ五員環の中間体(A3 の下図参照)にも確証はありません。この中間体からどのような経路でC-C結合が切れ、オゾニドに至るのかは、いろいろな推論はされていますが、直接の実験的な証拠はありません。次の回答にそうした推論の一つが示されています(詳しくは稲本「反応論による有機化学」(実教)などを参照して下さい)。
A3. オゾンの非局在化の式を描くと下のようになります。
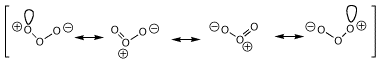
これとアルケンとの反応は、他の類似の試薬の反応(1,3-双極付加環化反応)からの類推で下のように考えられています。
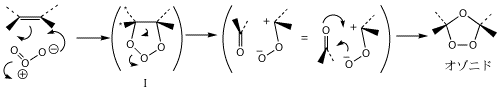
中間体Iに至る反応ではアルケンがπ電子をオゾンに与える形になりますが、このときにO-O単結合が弱くなることはありません(この点が過酸の場合と違います)ので、最初の段階でエポキシ環ができることはなさそうです。しかし、中間体Iから下図のような経路の反応がどうして不利なのかを理解するのは容易ではないと思います。
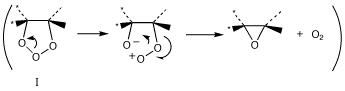
上のオゾニドに向かう経路と比較するとき、考えられる可能性は、
(i) Iの次の中間体では不安定な−O+が生じている、
(ii) 酸素分子の生成に伴うエネルギー程度では、ひずみの大きいエポキシ環の生成の駆動力としては不十分、
などです。
A4. 遊離基(free radical)の略称。共有結合(単結合)の左右の部分(原子または基)がそれぞれ電子を一個ずつもつように結合が切れたときに(ホモリシス)生成する不対電子をもった中性の化学種。一般に不安定で反応性に富む。
[芳香族求電子置換反応]
A1. 活性化基が複数ある場合には、もっとも強い活性化基で決まります。同じ基が複数ある場合など、条件が同じで位置が特定できない場合には、可能性のあるすべての生成物ができます。
A3. もっともな疑問です。
まず誤解している点から。反応のエネルギー図は、反応物(ベンゼン+臭素)と生成物(ブロモベンゼン+臭化水素)のエンタルピーを比較しているのであり、ベンゼンとブロモベンゼンではありません。
これは、熱力学データに基づいて議論するときに注意しなくてはならない点の一つです。ベンゼンとブロモベンゼンは異性体ではないので、熱力学的な比較により安定性を議論するのは困難です。
また、マクマリーの当該箇所には「ベンゼンよりもブロモベンゼンのほうが安定である」とは書いていません。「正確に引用する」ことは、引用するときにもっとも注意すべき点の一つです。多くの議論のすれ違いは、真意を誤解した(あるいは「ねじ曲げた」)引用に端を発しています。
次に、安定かどうかは、「何に対して」を明記しないと意味がありません。たとえば、熱には安定でも水とは反応することがあります。生成熱が大きいからといって、より安定であるとは限りません。
確かにBrが誘起効果でπ電子も引っ張りますので、いわゆる「ベンゼン環の非局在化エネルギー」だけを考えれば、共鳴による安定化は小さくなります(しかし、求電子置換反応に対しては安定になります)。しかし、同時にC-Br結合は(飽和のCに結合した場合と違って)π電子の非局在化により部分的に二重結合性をもちますので、(飽和のCに結合した場合よりも)結合解離エネルギーは大きくなっています。
最後に、問題のエンタルピー差(発熱反応)にもっとも大きく寄与しているのは、おそらく臭素分子と臭化水素分子との生成熱の差でしょう(化学便覧で調べればたぶんわかります)。
A4. (理学部化学科の大学院入試でも決して出題されません。)おそらく亜硝酸の起こす他の反応からの類推だと思われますが、次のような機構が提案されています[P.
Sykes(久保田訳)「有機反応機構・第5版」(東京化学同人)129ページ]。
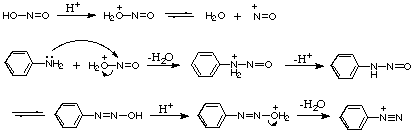
[カルボニル化合物の反応]
A1. どちらも同じように考えることができます。ヒドロキシルアミンとの反応も含めて、下に記しておきます。
Acid-catalyzed Reaction with R-NH2 (Primary
amines, oximes, and hydrazines): Addition-Elimination Mechanism
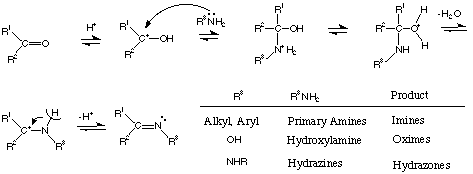
A2. いい質問です。確かにこちらも一部はプロトン化されます(酸塩基平衡)。しかし、上の反応でプロトン化されていない原料が消費されると、この速い可逆反応はプロトン化とは逆の方向へと進行します。
実際、多くの反応系では、反応の進行とは無関係のいくつもの化学平衡が生じています。カルボニル化合物の反応のような多段階反応の場合はこれが特に顕著であり、反応経路は決して一本道ではありません。自分で反応を考えるときにうまくいかないのは、(a)反応の進行に必要な化学平衡に気がつかない場合(対策:見落としている経路がないか探す)と、(b)反応の進行に無関係な化学平衡にとらわれている場合(対策:袋小路に陥ったら戻って別の経路を探す)とがあります。
迷路を解くのに似ていますが、袋小路にはちゃんと目印があります。それが見分けられるようになれば、反応生成物に到達するのが容易になるでしょう。
[酸化反応]
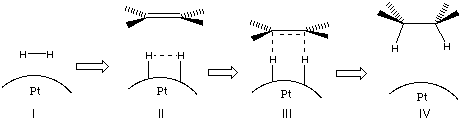
A1. どちらの条件下でも働く場合には、一般に酸性下のほうが酸化力が強いです。酸化剤は一般に電子が不足していますが、酸性下では酸化剤あるいはこれから生じる反応中間体にH+が結合し、電子不足の状態を生じやすいと考えればいいと思います。
A2. この反応では、水素分子は白金などの触媒となる金属の表面に吸着し、半ば解離して白金と結合したような状態になります(下図I→II、化学吸着といいます)。これにアルケンが平面の一方から接近し、白金表面の水素と結合します(下図II→III)。このため、cis付加の生成物が得られます(下図IV)。